Familial Hypercholesterolemia Treatment
Dec 08, 2022
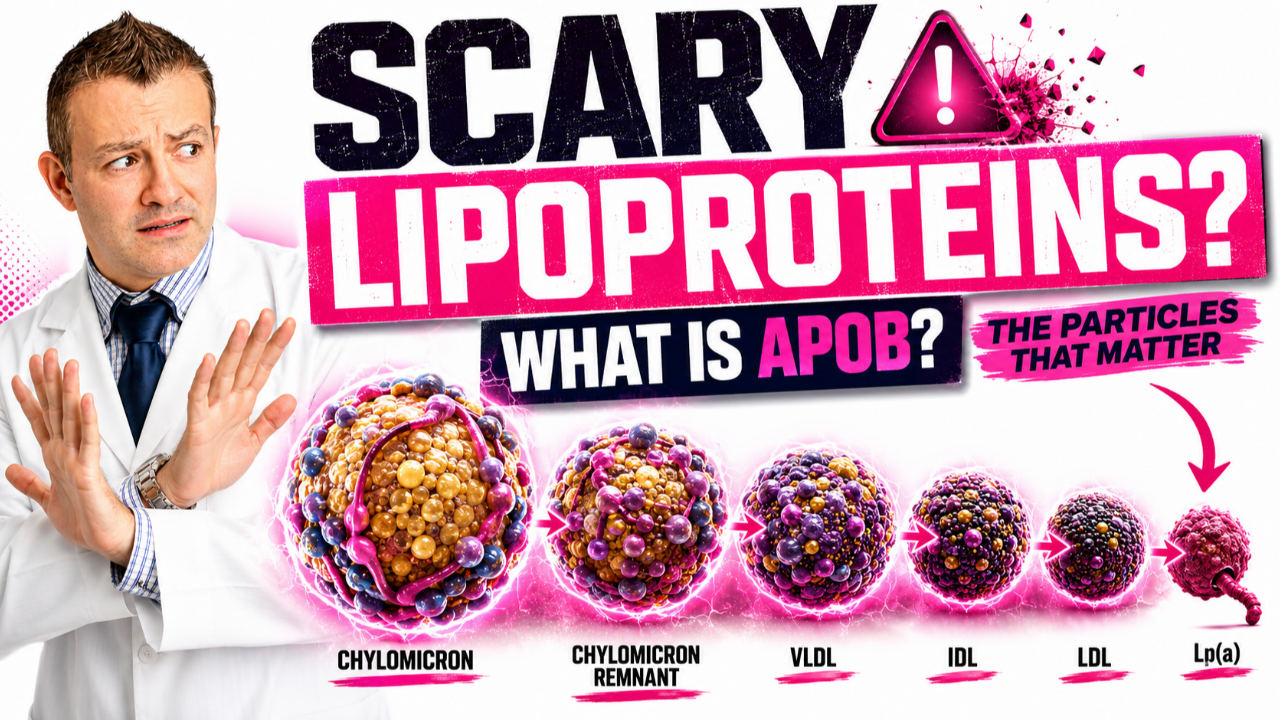
Familial Hypercholesterolemia
Familial hypercholesterolemia is a genetic disorder that gives people very high cholesterol and very early heart disease. It's not unusual to see teenagers with cholesterol levels in the 300-700 range.
Familial hypercholesterolemia can be subdivided into many different categories and classifications, and some are worse than others. Some don't respond as well to therapy.
Let's take a deep dive into this very dangerous genetic disorder.
Infographic Summary:

Difference Between Polygenic Hypercholesterolemia and Familial Hypercholesterolemia (Monogenic)?
Monogenic usually means you have one single gene mutation. Polygenic means you have multiple gene mutations.
How do you know if you have true familial hypercholesterolemia (which is monogenic) or polygenic hypercholesterolemia? They are different and confer different risks and lifespans and need to be treated differently based on how aggressive they are. Both can be very dangerous.
Polygenic hypercholesterolemia is due to mutations in many different genes, hence the name polygenic. Polygenic hypercholesterolemia is usually present in about 4.9% of the population. They are at increased CVD risk, but not as bad as monogenic (FH).
Monogenic hypercholesterolemia is usually due to a single gene mutation. This is what we normally call familial hypercholesterolemia. This is generally present in about 0.57% of the population, and these individuals are also at increased CVD risk.
Monogenic increases the risk of major adverse cardiac events (MACE) by 93%, whereas polygenic increases MACE by 26%. Both monogenic and polygenic conferred much higher risk than those with elevated cholesterol with no known underlying genetic mutations.
Read the full article;
https://jamanetwork.com/journals/jamacardiology/fullarticle/2760785
Heterozygous Familial Hypercholesterolemia
Heterozygous FH is when you have only one of the alleles, or one gene. Usually this comes from one parent. Homozygous FH is when you have two alleles (two genes) inherited from two parents. The homozygous is worse and usually gives higher LDL and cholesterol levels and confers much greater CVD risk. More on this later.
Homozygous Familial Hypercholesterolemia
This is when you inherit both genes (two genes), one from each parent. This is a more severe and dangerous form of FH. Most of these patients usually die before the age of 20. Below you can read a case of a 5 year old boy who had this and ultimately passed away.
Diagnosis
Usually this is diagnosed when a routine fasting lipid panel comes back with an LDL of 190 or higher. This is the easiest way to know that you may have this and needed it treated quite aggressively. People on ketogenic, high saturated fat diets need to come off that diet first, then recheck their lipids. A ketogenic diet will increase LDL levels up to 190 or more sometimes and we need to know if you really have familial hypercholesterolemia or not.
Later on we will discuss how you can figure out if it is monogenic or polygenic FH based on a questionnaire and a scoring system.
Symptoms
Unfortunately, often times there are no symptoms unless it's too late. If you have a more severe form, at a younger age, we can sometimes see some signs on your skin, eyes, and tendons.
People with familial hypercholesterolemia have very high levels of low-density lipoprotein (LDL) cholesterol in their blood. LDL cholesterol is known as "bad" cholesterol because it can build up in the walls of the arteries, making them hard and narrow and causing heart attacks and strokes. We now know that LDL directly causes atherosclerosis.
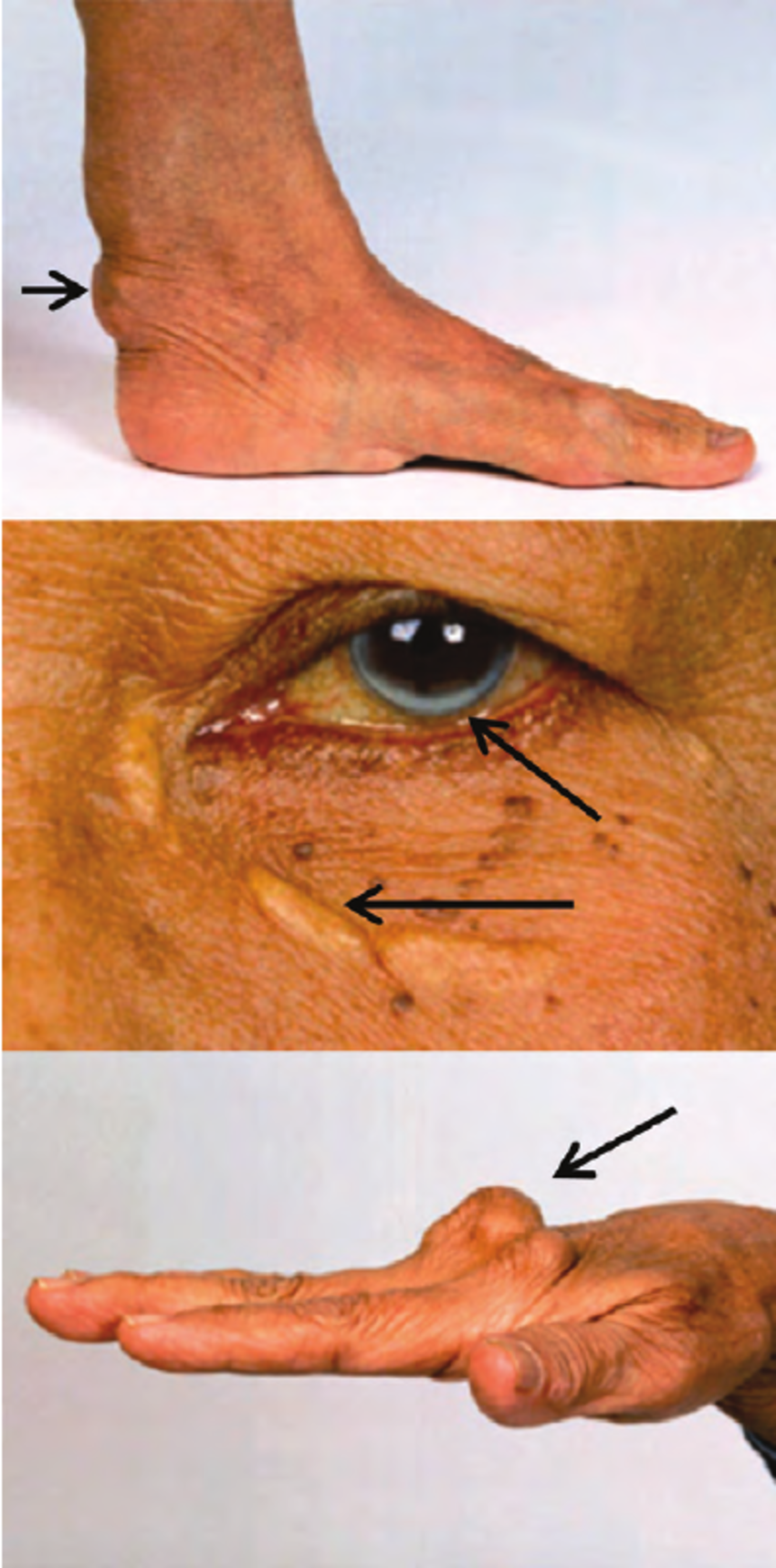
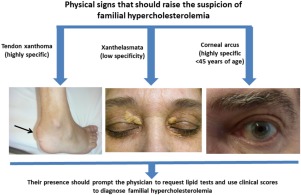
This excess LDL cholesterol can be deposited in various unusual locations; skin, some tendons, and around the iris of the eyes.
- Skin: Xanthomas. These are called xanthomas. The most common spots for cholesterol deposits to occur is on the hands past the knuckles, elbows and knees. They also can occur in the skin around the eyes. See below.
- Tendons: Xanthomas. Cholesterol deposits may thicken the Achilles tendon, along with some tendons in the hands. See the picture below. These are called xanthomas.
- Eyes: Arcus and Xanthelasmas. In the skin around the eyes you see light patches and deposits, these are called xanthelasmas . Inside the iris or colored part of the eye, you see a white arc. This is called arcus. High cholesterol levels can cause corneal arcus, a white or gray ring around the iris of the eye. This happens most commonly in older people, but it can occur in younger people who have familial hypercholesterolemia.
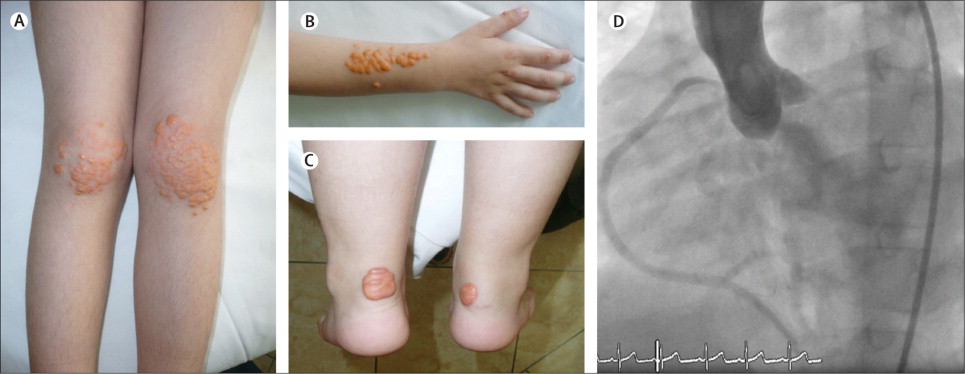
Very Young and Severe Homozygous Familial Hypercholesterolemia
Below are pictures from a 5 year old boy with Homozygous Familial Hypercholesterolemia. He had a cardiac event and was admitted to the hospital for work up and treatment. His right coronary artery needed a stent.
This case was described in The Lancet (https://www.thelancet.com/journals/lancet/article/PIIS0140673611614761/fulltext)
LDL - Apheresis
The young boy ended up having a special kind of blood filtration called an LDL-apheresis where they filter the LDL out of your blood stream like they do in dialysis. He died three months later of another acute ischemic cardiac event.
You can see the various cholesterol deposits all over his skin. You see this usually on the patellar tendon (knee) and achilles (heel).
As you can see, homozygous FH is the worst form of FH.
Treatment for Familial Hypercholesterolemia (monogenic)
Of course you want to start with lifestyle modification. Start with my 11 Ways to Lower Your Cholesterol Naturally article. It's important to start medications immediately alongside lifetyle modification due to how severe monogenic FH can be.
Generally the treatment is to start with the statins. I normally start with Crestor (rosuvastatin) 40 and add or adjust from there. You want to start the maximally tolerated statin dose. Crestor is the most potent and should provide good LDL lowering. All statins are generic and cheap now. Crestor also has the lowest incidence of side effects and is generally considered a "very clean" statin.
Next you would want to add ezetimibe (Zetia) to help further reduce LDL. You can usually reduce LDL by about 40-50% with this combination, depending on the person. This is also generic at this time.5Next we would add bile acid sequestrants. These help you sequester LDL receptors and eliminate them. The most commonly used one is cholestyramine, colesevelam, and colestipol. These are all generic.
If you need further LDL reduction, you will want to add evolocumab (Repatha). This is a twice a month injection that can provide an additional 65% reduction on top of the above. This is not currently generic and is expensive.
For about 80% of patients this is enough and will get them down into range.
The next step would be an LDL apheresis, but this is seldom needed.
Treatment for Polygenic Familial Hypercholesterolemia
Treatment options for polygenic hypercholesterolemia include cholesterol-lowering lifestyle changes and then adding medications as above. You usually have time in this scenario. I give patients 3 months to make lifestyle changes and then retest their lipid panel and see the effects.
Then you can start adding many of the above medications in that same order. Retest every 4-5 weeks to test the effects and see if you need to add more. In some cases you may have to start more than one at a time.
Life Expectancy
If discovered early enough, most people will live a normal life with normal life expectancy. If not treated at a young age, most men and women will live to about 45-55 with major complications.
If you have the homozygous version, as the 5 year old boy above had, most will only live to about age 20. It's imperative to screen all children at a young age.
Screening
It's important to screen all siblings, offspring, and parents once you have identified a patient with familial hypercholesterolemia. It is autosomal dominant, which means that if one parent carries the gene, about 50% of their offspring will have the gene.
Inheritance
Familial Hypercholesterolemia is autosomal dominant. That means if one parent has one gene, about 50% of their offspring will have it.
Incidence of Familial Hypercholesterolemia
Usually, this is seen in about 1 in every 200-250 people in the United States. Globally it's estimated at about 1 in every 311 persons. As you can see, this is quite common. (https://pubmed.ncbi.nlm.nih.gov/26976914/, https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.119.044795 )
How can you determine the difference between Polygenic Hypercholesterolemia and Familial Hypercholesterolemia?
Familial hypercholesterolemia is usually due to a genetic mutation in one gene, whereases polygenic, is due to multiple gene mutations. The risk of heart disease is usually much worse with Familial Hypercholesterolemia (monogenic).
There is something called the Dutch Lipid Score to help us figure out the difference between PH and FH.
Dutch Lipid Score
FORMULA
|
Criteria |
Points |
|
Family history |
|
|
First-degree relative with known premature* coronary and vascular disease, OR First-degree relative with known LDL-C level above the 95th percentile |
1 |
|
First-degree relative with tendinous xanthomata and/or arcus cornealis, OR Children <18 years with LDL-C level above the 95th percentile |
2 |
|
Clinical history |
|
|
Patient with premature* coronary artery disease |
2 |
|
Patient with premature* cerebral or peripheral vascular disease |
1 |
|
Physical examination |
|
|
Tendinous xanthomata |
6 |
|
Arcus cornealis prior to age 45 years |
4 |
|
Cholesterol level, mmol/L (mg/dL) |
|
|
LDL-C ≥8.5 (330) LDL-C 6.5 - 8.4 (250 - 329) LDL-C 5.0 - 6.4 (190 - 249) LDL-C 4.0 - 4.9 (155 - 189) |
8 5 3 1 |
|
DNA analysis – functional mutation LDLR, apoB and PCSK9 |
1 |
* Premature = <55 years in men; <60 years in women.
Score interpretation:
| Stratification | Total Score |
|---|---|
| Definite familial hypercholesterolemia | >8 |
| Probable familial hypercholesterolemia | 6-8 |
| Possible familial hypercholesterolemia | 3-5 |
| Unlikely familial hypercholesterolemia | <3 |
EVIDENCE APPRAISAL
The Dutch Criteria were constructed by the Dutch Lipid Clinic Network to identify patients with familial hypercholesterolemia based on known risk factors.
If you want the data and original article, see below.
Scientific Literature
CLINICAL PRACTICE GUIDELINES
OTHER REFERENCES
Al-Rasadi K, Al-Waili K, Al-Sabti HA, et al. Criteria for Diagnosis of Familial Hypercholesterolemia: A Comprehensive Analysis of the Different Guidelines, Appraising their Suitability in the Omani Arab Population. Oman Medical Journal. 2014;29(2):85-91. doi:10.5001/omj.2014.22.Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004. Sep;160(5):407-420 10.1093/aje/kwh236National Lipid Association Clinical Lipidology Resource Center
Grab my Actual Weight Loss book for more leading edge weight loss and heart health advice!
Grab my Mediterranean Diet Calorie Based Weight Loss Cookbook!
Join my Heart Healthy Community to discuss personalized health advice.
Still Have Questions? Stop Googling and Ask Dr. Alo.
You’ve read the science, but applying it to your own life can be confusing. I created the Dr. Alo VIP Private Community to be a sanctuary away from social media noise.
Inside, you get:
-
Direct Access: I answer member questions personally 24/7/365.
-
Weekly Live Streams: Deep dives into your specific health challenges.
-
Vetted Science: No fads, just evidence-based cardiology and weight loss.
Don't leave your heart health to chance. Get the guidance you deserve. All this for less than 0.01% the cost of health insurance! You can cancel at anytime!
[👉 Join the Dr. Alo VIP Community Today]
Cardiology & Obesity Medicine